

Sanger DNA Sequencing: Troubleshooting
It is part of our standard operating procedure that all sequence data generated at our Core facility are carefully inspected before they are made available to the end user. We have implemented rigorous process controls and a series of appropriate tools that enable us to immediately identify process- and/or instrument-related failures. In these very rarely occurring cases, all affected samples will be automatically resequenced, at no extra charge for our customers.
Should there be any issues with your sequencing data, we strongly recommend reviewing our compilation of the most common problems experienced in ABI Sanger sequencing, including example chromatogram images and descriptions of possible causes and solutions (see below).
Understanding and solving failed sequencing reactions and/or problematic sequencing data require thorough analysis of the corresponding sequencing chromatograms. These trace files are labeled with the extension .ab1 and can be viewed with one of the free software programs listed here.
If you would like assistance in analyzing your sequencing data, please contact us.
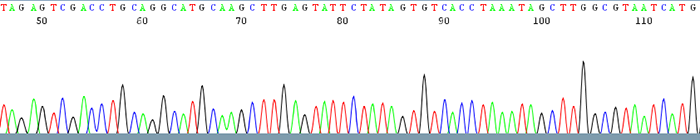
Good Quality Sequencing Data
1. Failed Sequencing Reactions (Sequence data contains mostly N's)
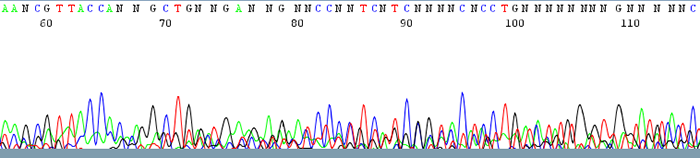
How to Identify:The trace is messy with no discernable peaks
What is the cause?It is difficult to pinpoint the exact reason as to why sequence reaction fails due to the fact that there is no good quality data to analyze.
Some of the more common reasons and how to fix them can be found here:
- The template concentration is too low.This is the number one reason as to why a sequence reaction fails. We ask for a template concentration to be between 100ng/µL and 200ng/µL. It is difficult to get an accurate reading of such a low concentration on a regular spectrophotometer so we recommend using an instrument such a Nano Drop that is designed to measure small quantities of DNA.Please note:It is very common to see samples with weak concentrations seem to give acceptable sequence data when sequenced one time and fail the next. This happens when the signal intensity is of borderline readability and data gets detected by the software only some of the time.
- Poor quality DNA.Make sure the DNA is of high quality and the 260 to 280 OD ratio is 1.8 or greater. Contaminants greatly hinder the ability to obtain good sequence data. Make sure to clean up DNA to get rid of excess salts, contaminants and PCR primers
- Too much DNA.Excessive amounts of template DNA can kill a sequence reaction
- Bad primer or incorrect primer added to template.Make sure to use a good quality primer and the priming site is located on the template strand. You can read more about primer design here
- Blocked capillary on the sequencer.This is a problem on our end and can randomly occur at any time. The occurrence rate is very low averaging one in every 200 reactions. We will happily rerun any sample that we think may have failed for this reason.
2. Chromatograms show a lot of noise or background along the bottom of the trace
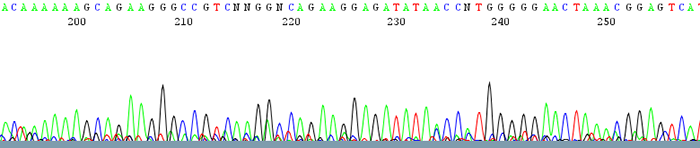
How to Identify:The trace has some discernable peaks but also has a lot of background noise running along the bottom that can interfere with base calling. The quality scores of the peaks are usually very low.
What is the cause?This is usually due to low sample signal intensity. Low signal intensity is a result of poor amplification due to either:
- Low starting template concentration.Make sure your template concentrations are between 100ng/µL and 200ng/µL
- Low primer binding efficiency.Make sure the primer has a high binding efficiency, is not degraded and does not have a large n-1 population. You can read more about primer design here.
- Any other reason listed under the failed reaction section can also not be ruled out.
3. Poor data is seen following a region of mononucleotides (single base)
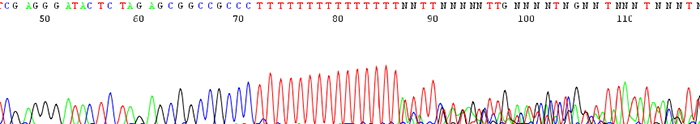
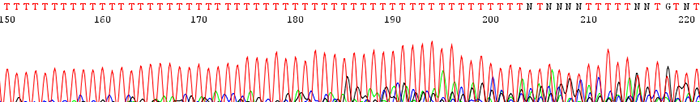
How to Identify:The sequence trace becomes mixed and unreadable after a stretch of mononucleotides. (After a run of a single base)
What is the Cause?The polymerase slips on the stretch of mononucleotides causing a disassociation and re hybridization in a different location. This causes varying sized fragments, creating a mixed signal after the region.
How do I fix the problem?There is currently no way to effectively sequence directly through such a region. A primer can be designed that sits just after the mononucleotide region or else a primer can be designed that sequences toward it from the reverse direction.
4. Good quality data that suddenly comes to a hard stop
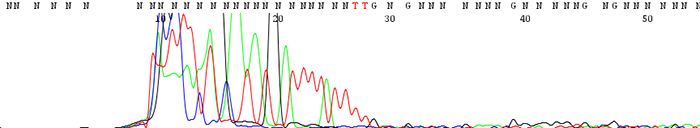
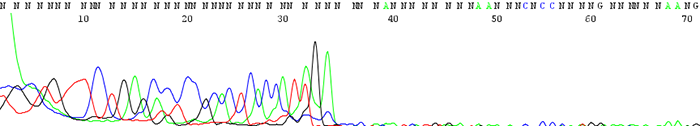
How to Identify:The sequence looks fine before it suddenly either terminates or the signal intensity drops dramatically.
What is the cause?This is usually a sign of secondary structure present in the template. Complementary regions fold up on themselves to form hairpin structures that the sequencing polymerase cannot pass through. Long stretches of Gs or Cs can also cause problems for the sequencing polymerase.
Important fact to note:If the secondary structure is weak enough, we often see a reaction successfully sequence through half of the time and terminate early the other half. Simply put, the polymerase successfully passes through the secondary structure only some of the time to give good sequence.
How can I fix the problem?We offer an alternate sequencing protocol that can sometimes help this situation. It is a different dye chemistry designed by ABI that helps to pass through secondary structure and other difficult templates. It is not guaranteed to work and should be tested on a few samples at first before using it on a large batch of samples. Many people find it helpful while some do not. It can be ordered by clicking on the "difficult template" button on the right hand side of the order form. Because of the extra reagents involved it costs a little bit more than a regular sequencing reaction. You can view the rates by clicking here. Please call us it you would like advice as to whether this protocol might be beneficial to you.Please note:This protocol will not help sequences that completely fail with the standard sequencing protocol. There must be visible signs in the chromatogram that will hint to a problem with secondary structure.
***An alternate method of sequencing a difficult region is designing a primer that either sits directly on the area of secondary structure, or designing a primer that avoids the region all together.
***Known regions to be problematic include sequences containing the LOXP site and the Att site of lambda phage used by Gateway entry vectors
5. Good quality data that turns to double sequence (2 or more peaks in the same location)
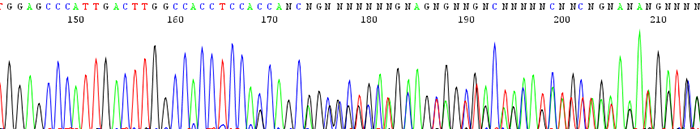
Good quality data that turns to double sequence (2 or more peaks in the same location)
How to identify:The sequence trace begins high quality and then becomes mixed.
What is the cause and how can I correct it?There can be a couple different causes of this.
- There is colony contamination and there are 2 or more clones being sequenced.If two colonies are accidently picked at the same time, you end up sequencing more than one insert. This can be avoided by making sure that only one colony is picked and sequenced.
- The DNA contains a toxic sequence.This can occur if the gene that was cloned is expressed in E. coli and is toxic to the cell. This happens most often in high copy vectors and templates with low G + C ratio. This can be avoided by using a low copy vector or by growing the cells at 30deg C. Also do not overgrow the cells which can cause more deletions or rearrangements in the desired sequence.
6. Double sequence that starts from the beginning of the trace (mixed template)
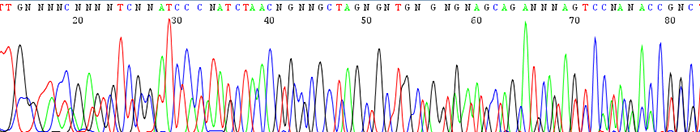
How to identify:The sequencing trace has two or more peaks in the same location. The quality scores for each peak are very low. The text file will contain a lot of N's
What are the causes and what can I do to fix them?There can be many different situations that give rise to this problem.
- There is more than one template present in the sequence reaction.Make sure to give us only one template per sequence run
- There was more than one primer added per template reaction.Be sure to add only one primer to one template tube. If you have one template to sequence in both the forward and reverse direction you should give us 2 tubes to sequence. One tube will contain the template and the forward primer; the other tube will contain the template and the reverse primer.
- There is more than one priming site on the template strand.Make sure that the template you are sequencing has only one priming site for the primer you are using
- The PCR reaction was not cleaned up properly before you sent it into us.Be sure to rid the pcr reaction of any residual salt and primers before you send it for sequencing. Most PCR purification kits work fine. We also have a PCR purification service where we purify PCR reactions in 96 well formats before sequencing. You can read more about this service here.
7. Sequence gradually dies out causing early termination in read length
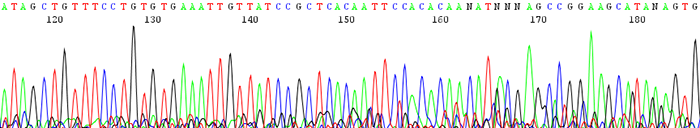
How to identify:The sequence starts out with high quality peaks and either stops prematurely or becomes messy downstream. The raw data shows very high signal intensity at the very beginning of the sequence and sharply decreases.
What is the cause and how can I fix the problem?The number one reason for this is having too much starting template DNA. The template becomes over amplified and uses up all of the florescence that we label the DNA with at the beginning of the sequence. This can be corrected by lowering your template concentration to between 100ng/µL and 200ng/µL. Use lower amounts for short pcr products under 400bp. View the concentration guide here.
8. Poorly resolved sequence traces where the peaks are very broad and not sharp or distinct
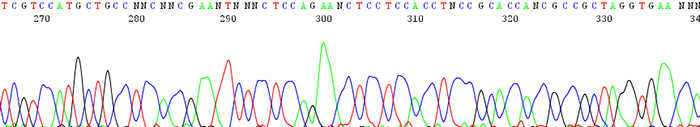
How to identify:The peaks are very broad and blobby instead of narrow and sharp.
What is the cause and how can I fix it?This is a very rare occurrence though seems to happen now and then with certain people's samples. Popular theory is that there is some kind of unknown contaminant in the DNA. People have noticed it mostly when using filter purification kits. Try another cleanup method or dilute the template.
This can also occur if the polymer in our sequencers starts to break down. We usually detect if this happens and if it does we see a loss of resolution in the data of all users on a run. We automatically rerun these samples before you ever see the data.
9. Large dye blob that occurs around 70 base pairs
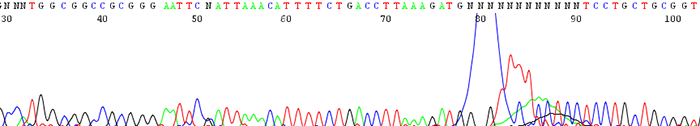
How to Identify:The sequence will contain a large dye blob that may or may not overshadow the regular base calling at around 70 base pairs.
What is the cause?This can be a result of one of a couple reasons.
- Dye blobs are thought to occur when the sequencing chemistry interacts with an unknown contaminate in the DNA. They become apparent especially when the signal intensity (rfu) signal is very low. This is not a common problem though we do notice that it occurs more frequently in certain individual's samples.
- Dye blobs can also happen if during our cleanup process we do not remove all of the unincorporated dye before we sequence the samples. We usually catch this since we will see that the vast majority of the samples in a particular run will have the problem. In this situation we rerun the samples before we send you the data and you don't see it.
10. The sequence starts out noisy or mixed and changes to good quality further down the trace
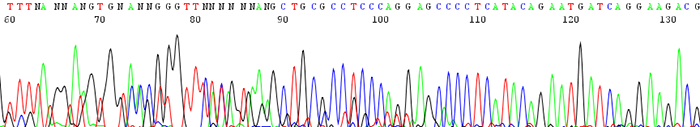
How to identify:There are many N's in the beginning of the sequence and the peaks appear noisy or mixed. The trace becomes clean with identifiable peaks downstream.
What is the cause and how to I fix it?This is usually the result of primer dimer formation. Primer dimers form when a primer self hybridizes due to complementary bases on the primer itself. Make sure your primer is not likely to amplify itself by having it analyzed by one of the many free primer analysis programs online. You can also view a primer design guide here.
11. Sharp signal spike that occurs randomly in the trace
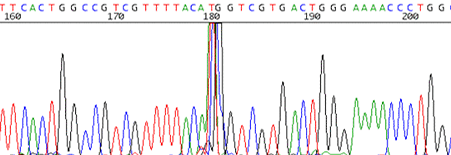
How to identify:The trace reads cleanly and is disrupted by a very sharp signal spike that spans about 2 or 3 base pairs.
What is the cause and how do I fix it?It is believed that these spikes are caused by small air bubbles that can get trapped in the capillaries of the sequencer. We will rerun the sample for you if the spike is in an area of particular interest for you.
12. Chromatogram shows an insertion or deletion (indel)
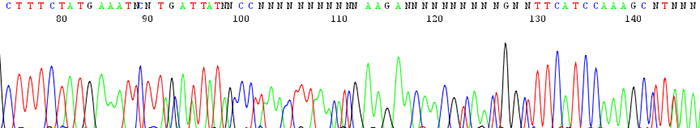
How to identify:The sequencing trace is good quality in the beginning and becomes mixed further downstream after an indel region. The peaks in the mixed region are often times shifted and a small peak can occur either before or after a larger one.
What is the cause and how do I fix it?An indel is caused by either an insertion or deletion on the template strand. They can be seen when sequencing PCR products with polymorphic regions. Polymorphisms can be a direct result of a heterozygous individual or because of a random mutation that occurred during the cloning process. Random mutations occur much more readily if cloned template is toxic or unstable in the E.coli host. If the mutation is caused by template instability, try cloning into a different strain or using a low copy vector. You can also try sequencing from the reverse direction.
13. The trace shows a delayed start in the first base of the read
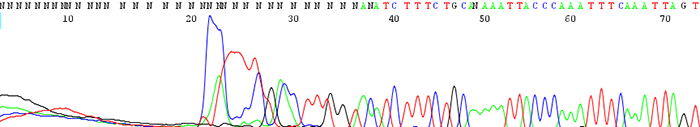
How to identify:The sequencing trace is good quality in the beginning and becomes mixed further downstream after an indel region. The peaks in the mixed region are often times shifted and a small peak can occur either before or after a larger one.
What is the cause and how do I fix it?An indel is caused by either an insertion or deletion on the template strand. They can be seen when sequencing PCR products with polymorphic regions. Polymorphisms can be a direct result of a heterozygous individual or because of a random mutation that occurred during the cloning process. Random mutations occur much more readily in the cloned template is toxic or unstable in the E.coli host. If the mutation is caused by template instability, try cloning into a different strain or using a low copy vector. You can also try sequencing from the reverse direction.
14. The peaks look fine in the chromatogram file but many of them are labeled as N's. The peaks may also blast out of range
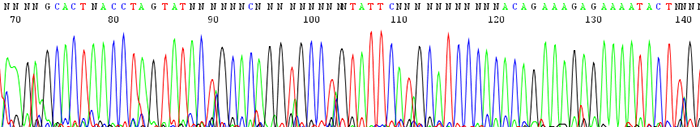
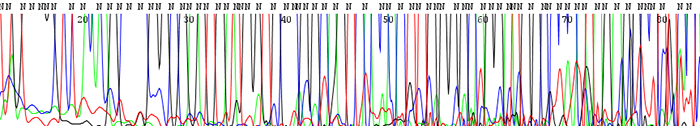
How to identify:The peaks look clear and distinct as depicted in the chromatogram file but the base caller marks them as Ns. This is particularly prevalent in the earlier portion of the sequence. The signal intensities are extremely high and much of the time the peaks can be accurately be named by eye.
What is the cause and how do I fix the problem?This is always caused by high starting template concentrations. The DNA is over amplified and the resulting signal gets blasted out of the range where the base caller on the sequencer can accurately read. We see it most often in short PCR products where it is easy to submit too much DNA. Make sure to keep template concentrations closer to 100ng/µL if the PCR products are under 500bp.