

A simple algorithm for detecting CRISPR variants from NGS reads
Here I outline a simple algorithm that will detect possible CRISPR variants from NGS reads. The algorithm is developed for paired-end NGS sequencing of a CRISPR sample that might contain several variants. The amplicons are between 200 bp and 300 bp, and we typically generate between 70,000 and 100,000 NGS reads for each sample (assuming the sample is of high quality and has the recommended DNA concentration). The following figure shows the results for a typical sample.
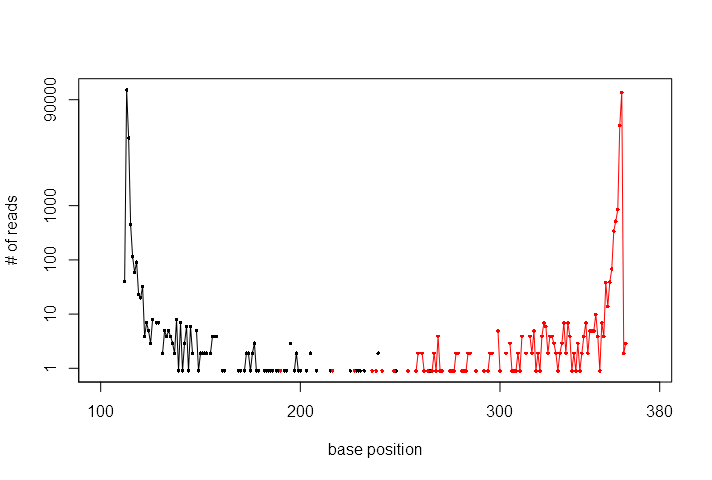
(The number of NGS reads matched to the reference sequences. Black dots represent reads that match in the forward direction while red dots represent those that match in the reverse direction).
The key point to notice is that, for such a short amplicon, the data is highly redundant. Although we have several hundred thousand reads, the number of unique sequences is actually much smaller. As the above figure demonstrates, most of the NGS reads have hundreds, if not thousands of exact copies. To make the following description simple, we have to make a clear distinction between "sequences" and "reads". Sequences are some abstract mathematical entities. "ATCGATCG"" is a sequence. "ATCGATCC" is another, a different sequence. There is only one sequence that is "ATCGATCG". Reads are what we get from the NGS process. For each read, there is exactly one corresponding sequence. Many different reads, however, can correspond to the same sequence.
The algorithm is as follows:
-
Filter
Filter out low frequency reads. The exact cutoff does not really matter much. A cutoff, c, somewhere between 10 and 100 should be fine. Only the HFR (high frequency reads) will be considered in the next step. -
Cluster
The HFR, by their very nature, correspond to a rather small set of sequences, the HFS (High Frequency Sequences). Usually we are left with only a few thousand sequences to work with. Sequences within this set are often similar enough that we can proclaim, with very high confidence that they originated from the same amplicon. For example, reads that form a ladder:ATCGATCG TCGATCGC CGATCGCCOr two sequences that are different at only one position, while the differences of their frequencies suggest that the low frequency one is the result of a sequencing error. We can cluster these sequences into an even smaller number of clusters, the HFSC (High Frequency Sequence Clusters). Each of these clusters contains one or more HFS. All the sequences are very similar to each other within the cluster. -
Pair
We examine all possible pairs of these HFSC. Remember that each sequence corresponds to tens or hundreds of reads, so each of these HFSC pairs corresponds to hundreds or thousands of possible pairs of NGS reads. We are only interested in those read pairs that are mate-pairs (i.e. one read for sequence s1, and the other read for s2 are from the same amplicon). Only cluster pairs with a large number of mate-pairs go to the next step. Any pair (c1, c2) that passes this step has more than pm NGS mate-pair reads, where pm can be somewhere between 10 and 100. -
Merge
We are almost done, but before we finish, there is one final step we have to go through. Each pair of these HFSC, by construction, corresponds to many NGS reads pairs. To build a final finished sequence for each of these pairs, we have to merge the forward and reverse reads. If the reference sequence is known, the process is straightforward. Without a reference, the merging could be risky. One obvious parameter to check is the overlap between the forward and reverse reads. If the overlap is significant (a 4bp overlap seems to work for most cases), we can simply merge them. Otherwise, we need to consider the possibility of gaps (i.e. the amplicon might be longer than the sum of two reads).
Questions to hwang12@mgh.harvard.edu