

Home >
AAV_Genome_Sequencing
>
current
AAV Genome Sequencing: Data Retrieval
Explanation of pipeline and delivered data files Our workflow is a scalable, modular pipeline for analyzing recombinant AAV (rAAV) sequencing data generated from Oxford Nanopore platforms. Based on Oxford Nanopore Technologies' EPI2ME AAV QC workflow, sequenced rAAV reads are processed through the following steps:
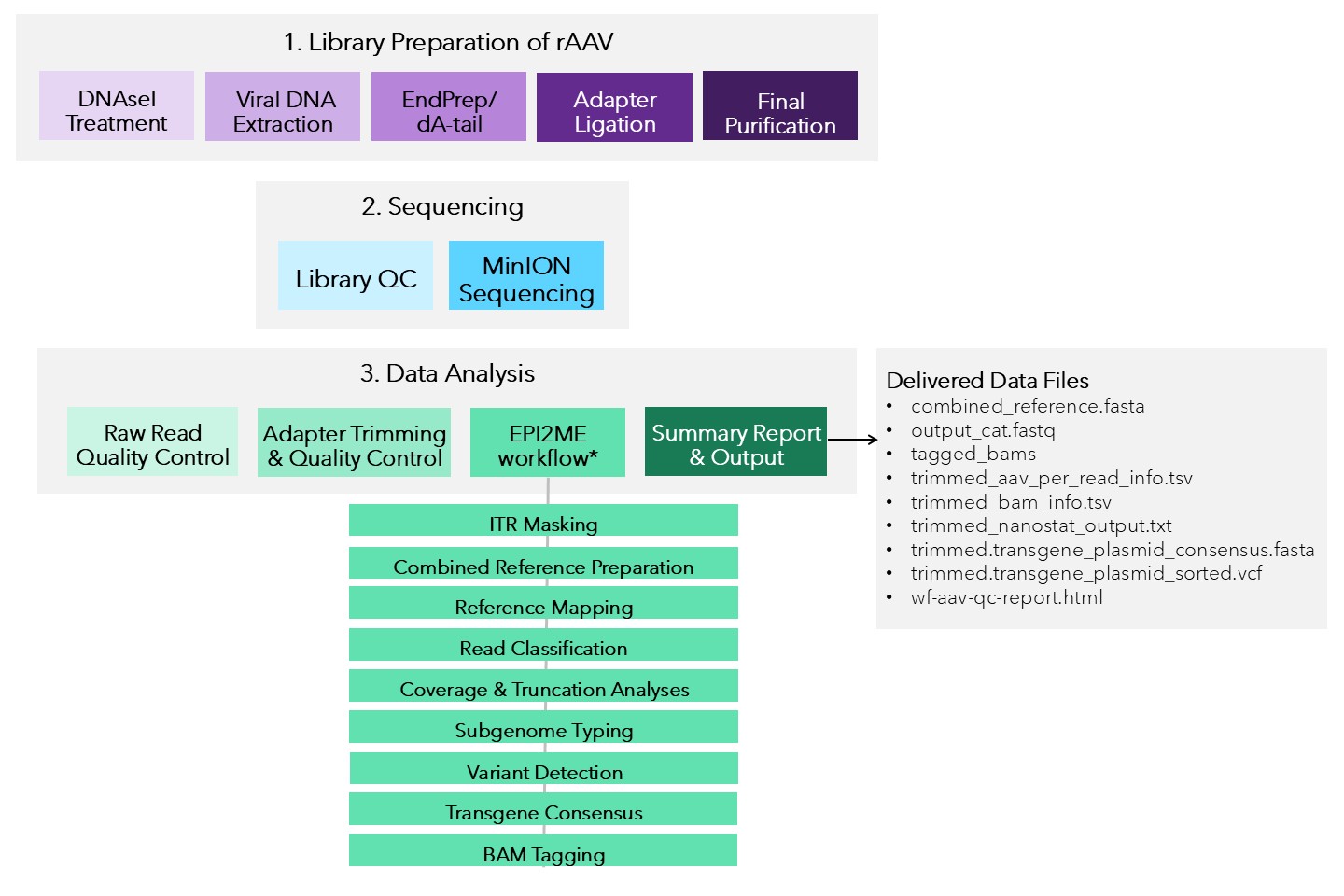
Figure 1. Overview of rAAV workflow from sample preparation, library construction, sequencing and analysis.
| Step | Functionality |
|---|---|
| Input Data | Accepts raw Nanopore FASTQ reads and per-sample metadata via a CSV file. |
| Raw Read Quality Control | Uses NanoStat to evaluate raw read quality metrics such as length, quality score, and yield. |
| Adapter Trimming & Quality Control | Removes adapter sequences and performs post-trimming QC to assess read data integrity and retention. |
| ITR Masking | Detects and masks variable Inverted Terminal Repeat (ITR) regions in the transgene cassette to improve alignment accuracy. |
| Combined Reference Preparation | Builds a combined reference by concatenating the host genome, masked transgene, Rep-Cap, and helper plasmids. |
| Reference Mapping | Aligns reads to the combined reference using minimap2. |
| Read Classification | Identifies the genomic source of each read (e.g., transgene, helper, host). |
| Coverage & Truncation Analyses | Calculates depth of coverage from ITR to ITR and detects common truncation hotspots. |
| Subgenome Typing | Categorizes reads into rAAV subgenome types (e.g., complete, partial, snapback, plasmid backbone). |
| Variant Detection | Identifies sequence variants in the transgene plasmid relative to the reference. |
| Transgene Consensus | Generates a polished consensus sequence of the transgene insert from all mapped reads. |
| BAM Tagging | Adds metadata tags to each read indicating genome source and subgenome classification. |
| Summary Report & Output | Produces interactive QC reports, read alignment statistics, variant summaries, tagged BAMS, and consensus FASTA. |
For a detailed breakdown of the workflow and visual diagrams of rAAV subgenome types, please visit the official EPI2ME AAV Workflow GitHub repository.
Delivered Data Files Nine data files will be generated per sample. These files are prefixed with the TUBE_ID provided in the sample submission form.
- prefix_combined_reference.fasta
- prefix_output_cat.fastq
- prefix_tagged_bams
- prefix_trimmed_aav_per_read_info.tsv
- prefix_trimmed_bam_info.tsv
- prefix_trimmed_nanostat_output.txt
- prefix_trimmed.transgene_plasmid_consensus.fasta
- prefix_trimmed.transgene_plasmid_sorted.vcf
- prefix_wf-aav-qc-report.html